
新闻资讯News Center
联系我们contact us
始终着眼于客户和社会的期望,
在他们需要的时候提供市场领先的服务。
新版欧盟《医疗器械法规(MDR)》今日执行 !
新版欧盟《医疗器械法规(MDR)》今日执行 !
2021年5月26日,对于全球医疗器械制造商来是一个特别的日子,经过为期一年的延期,终于在各界关于其是否会再次延期的猜测中,迎来了强制生效的日期,至此,MDD已经正式退出历史舞台。
MDD
MDD是欧盟医疗器械93/42/EEC指令的简称,适用于在欧盟全体成员国医疗器械的的准入要求,属于强制认证,自1994年落地起强制实施到2021年5月25日正式结束,MDD完成了其长达二十几载的服役周期。

MDR
欧盟医疗器械法规MDR(REGULATION (EU) 2017/745)于2017年5月5日定稿,同年5月26日正式生效。欧盟委员会于2020年4月17日通过关于MDR实施日期推迟一年的建议,MDR生效日期推迟至2021年5月26日。
新的法规将替代原有的2个指令:医疗器械指令 (MDD 93/42/EEC)、有源植入性医疗器械指令 (AIMD 90/385/EEC)。新法规在受监管方、参与监管方、产品分类、符合性评估途径、临床监管、欧盟医疗器械数据库的使用等方面都做出了重大调整,提出了很多新的、更高的要求。已通过MDD/IVDD指令CE认证的产品,也需要重新评估后才能获得MDR/IVDR CE证书。虽然新法规给企业4年的过渡期,但实际企业还是有有很多转换工作需要执行。此次重大变更,在欧盟医疗器械历史上具有“革命性”的意义。如何才能满足新法规的要求,已成为中国制造商面对的巨大挑战。

一、 新法规下欧盟医疗器械的主要分类
根据欧盟新颁布的医疗器械法规《医疗器械法规》(2017/745,MDR)和《体外诊断器械法规》(2017/746,IVDR),欧盟将医疗器械分为两大类别:医疗器械MD和体外诊断器械IVD。MDR法规执行时间为2021年5月26日,IVDR法规执行时间为2022年5月26日。
侵入式医疗器械MD根据风险等级再细分为 I、IIa、 IIb、III类;非侵入式体外诊断器械IVD依据风险等级由低到高细分为A、B、C、D四类。
二、 新欧盟医疗器械法规MDR的主要变化
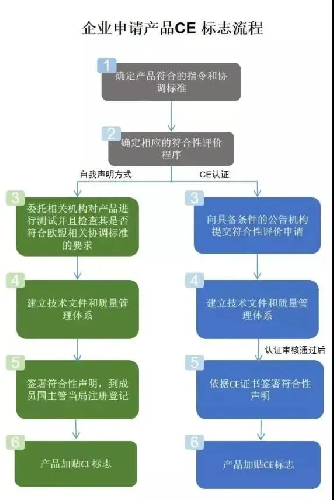
总体来说,欧盟MDR新规更加关注临床性能、更好的医疗器械可追溯性和对患者更大的透明度,具体变化有:医疗器械的范围扩大;提出医疗器械新概念和定义;设立中央电子资料库(Eudamed);设立产品独立的产品识别码(UDI);完善了医疗器械的通用安全和性能要求;加强对技术文件的要求;加强器械上市后的监管;完善临床评价相关要求;对授权认证机构(NB)提出严格要求等,这意味着对进入欧洲市场的医疗器械将实施更严格的限制,对行业企业提出了更高的要求。
三、 相关企业应该做好应对准备
欧盟MDR新规将给中国出口企业带来成本增加、认证周期拉长和合规风险增加等问题,建议相关企业做好相应准备。一是加大MDR新规关注力度并做好CE认证更新。过渡期前签发的CE证书有效期不超过5年,并于2024年5月27日失效,而2021年5月26日后申请CE认证的,必须按照MDR法规要求办理,并由授权NB签发;二是树立质量意识和责任意识,按照MDR法规要求合规生产,保障产品和标准的符合性;三是与国外客户加强联络沟通,明确生产标准和认证要求以避免后续认证和价格纠纷。
中易证提供医疗器械MDR认证和欧盟授权代表服务,如需要了解这方面信息,可和我司工作人员联系:021-5298370

 销售一部
销售一部
